
 Imprimir
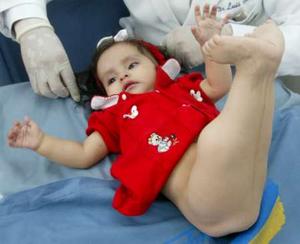
ImprimirEl neurofibroma es una lesión de origen desconocido que puede ocurrir en el nervio periférico, en los tejidos blandos, en piel o hueso. se caracteriza por la presencia de elementos nerviosos periféricos y tejido conectivo dispuestos en una forma difusa y desordenada. Normalmente ocurre como una lesión solitaria más que como lesión múltiple (como es la neurofibrosis). Los neurofibromas que ocurren como la parte de la enfermedad de Von Recklinghausen son generalmente más grandes que las lesiones solitarias y tienen un 4% de probabilidades de transformación maligna. Las lesiones solitarias, generalmente, se presentan a la edad entre 20 a 30 años y a menudo se levanta como una masa superficial indolora en la dermis. Los neurofibromas se pueden formar también en el plexo braquial, plexo lumbosacro, nervios periféricos o nervios espinales.
Cuando se presenta como lesión múltiple hablamos entonces de neurofiromatosis (NF) que son trastornos genéticos del sistema nervioso que causan el crecimiento de tumores no cancerosos a lo largo de los nervios y también pueden provocar anomalías en la piel y huesos.Existen dos formas principales de NF: la NF1 y la NF2.
FRECUENCIA CON LA QUE SE PRODUCEN LAS NF
La NF1 es uno de los trastornos genéticos más comunes que existen. La NF2 es menos común. Ambas manifestaciones de NF ocurren en todos los gurpos étnicos y raciales del mundo y afectan a ambos sexos pro igual.
CAUSAS
Son causadas por dos genes anormales distintos. El gen de la NF1 se encuentra en el cromosoma 17 y el gen de la NF2 en el cromosoma 22. El gen anormal de cada forma de NF es dominante autosomático, por lo que cada hijo de un padre con NF (suponiendo que el otro padre no está afectado) tiene una probabilidad del 50% de heredar el gen de la NF.
SÍNTOMAS
NF1: La NF1 presenta siete síntomas típicos y se diagnostica en aquellas personas que presentan dos o más de los siguientes síntomas: -Seis o más manchas de color marrón claro en la piel, conocidad como manchas "café con leche". Se encuentran presentes al nacer o aparecen antes de los dos años de edad. Pueden aumentar de tamaño y cantidad u oscurecerse.
-Pecas que aparecen en las axilas o la zona de la ingle antes de los siete años de edad.
-Dos o más neurofibromas debajo de la piel o a mayor profundidad que crecen sobre los nervios y por lo general se desarrollan cerca de la pubertad.
-Un tumor en el nervio óptico (glioma óptico).
-Dos o más diminutos nódulos de Lisch de color marrón claro u oscuro, que son pequeñas acumulaciones de pigmento que aparecen en el iris pero no causan problemas en la vista.
-Una variedad de defectos en los huesos, como curvatura de las piernas por debajo de la rodilla.
-Antecedentes familiares de NF1 en un padre, hermano o hijo.
NF2: En la mayoría de las personas afectadas se caracteriza por la formación de tumores en el nervio que conecta el oído con el cerebro. Estos tumores generan presión sobre los nervios acústicos y producen la pérdida de la audición. También se pueden desarrollar tumores en tejidos nerviosos de otras partes del cuerpo y un tipo especial de catarata en una etapa temprana de la vida.
DIAGNÓSTICO
NF1: Se diagnostica mediante un examen físico que puede constar de distintas pruebas como radiografías, tomografías computarizadas y resonancias magnéticas... En algunos casos también se requieren pruebas genéticas de una muestra sanguínea para confirmar el diagnóstico. En niños menores de 8 años de edad se pueden presentar manchas de café con leche sin tener ningún otro síntoma de NF1. Se recomiendo entonces realizar un seguimiento de estos niños por si aparece cualquier otro síntoma de trastorno.
NF2: Por lo general se diagnostica mediante un examen físico y el médico puede recomendar una serie de pruebas e imágenes como la resonancia magnética para obsevar el cerebro y la médula espinal y así poder detectar tumores muy pequeños y permitir su diagnóstico temprano. También se realizan, en algunos casos, pruebas genéticas de una muestra sanguínea, así como pruebas de audición y acudir al oftalmólogo para detectar cualquier síntoma de cataratas u otros problemas oculares que puedan llevar a la pérdida de la vista.
TRATAMIENTO
NF1: No hay cura para la NF1 pero se pueden tratar algunos de sus efectos. Se pueden extirpar los tumores de la piel que causan dolor o desfiguración mediante cirugía, pero estes con frecuencia vuelven a crecer.NF2: Tampoco existe ninguna cura para este tipo pero se puede realizar cirugía y radioterapia para ayudar a controlar los síntomas. Las resonancias magnéticas pueden detectar pequeños tumores, lo que permite un tratamiento temprano. En algunos casos la extirpación de parte de un tumor seguida de radioterapia puede ayudar a aliviar los síntomas.
LA HISTORIA DE HUANG CHUNCAI, EL HOBRE ELEFANTE
Huang Chuncai sufre desde pequeño un neurofibroma en la cara, un tumor que crece poco a poco sin parar. Hasta los 31 años que tiene actualmente tuvo que vivir con un bulto en la cara que crecía y crecía hasta llegar a pesar en la actualidad 15 kgs. Desde pequeño sufrió el desprecio de sus compañeros, por lo que abandonó la escuela cuando tenía 10n años. Desde entonces vivió encerrado. Él afirma que no tuvo tan sólo un día feliz en su vida. Pero ahora su vida puede cambiar al someterse a una cirugía. Los especialistas aseguran que se trata de un caso complicado porque el tumor es enorme y hay demasiados vasos sanguíneos dentro, lo que hace que la operación sea muy difícil y si no pueden parar la sangre durante la intervención Huang morirá. Pero a pesar del riesgo Huang está decidido a sacrificarse porque asegura que nunca ha podido disfrutar de un día feliz.
cara, un tumor que crece poco a poco sin parar. Hasta los 31 años que tiene actualmente tuvo que vivir con un bulto en la cara que crecía y crecía hasta llegar a pesar en la actualidad 15 kgs. Desde pequeño sufrió el desprecio de sus compañeros, por lo que abandonó la escuela cuando tenía 10n años. Desde entonces vivió encerrado. Él afirma que no tuvo tan sólo un día feliz en su vida. Pero ahora su vida puede cambiar al someterse a una cirugía. Los especialistas aseguran que se trata de un caso complicado porque el tumor es enorme y hay demasiados vasos sanguíneos dentro, lo que hace que la operación sea muy difícil y si no pueden parar la sangre durante la intervención Huang morirá. Pero a pesar del riesgo Huang está decidido a sacrificarse porque asegura que nunca ha podido disfrutar de un día feliz.
-Pecas que aparecen en las axilas o la zona de la ingle antes de los siete años de edad.
-Dos o más neurofibromas debajo de la piel o a mayor profundidad que crecen sobre los nervios y por lo general se desarrollan cerca de la pubertad.
-Un tumor en el nervio óptico (glioma óptico).
-Dos o más diminutos nódulos de Lisch de color marrón claro u oscuro, que son pequeñas acumulaciones de pigmento que aparecen en el iris pero no causan problemas en la vista.
-Una variedad de defectos en los huesos, como curvatura de las piernas por debajo de la rodilla.
-Antecedentes familiares de NF1 en un padre, hermano o hijo.
NF2: En la mayoría de las personas afectadas se caracteriza por la formación de tumores en el nervio que conecta el oído con el cerebro. Estos tumores generan presión sobre los nervios acústicos y producen la pérdida de la audición. También se pueden desarrollar tumores en tejidos nerviosos de otras partes del cuerpo y un tipo especial de catarata en una etapa temprana de la vida.
DIAGNÓSTICO
NF1: Se diagnostica mediante un examen físico que puede constar de distintas pruebas como radiografías, tomografías computarizadas y resonancias magnéticas... En algunos casos también se requieren pruebas genéticas de una muestra sanguínea para confirmar el diagnóstico. En niños menores de 8 años de edad se pueden presentar manchas de café con leche sin tener ningún otro síntoma de NF1. Se recomiendo entonces realizar un seguimiento de estos niños por si aparece cualquier otro síntoma de trastorno.
NF2: Por lo general se diagnostica mediante un examen físico y el médico puede recomendar una serie de pruebas e imágenes como la resonancia magnética para obsevar el cerebro y la médula espinal y así poder detectar tumores muy pequeños y permitir su diagnóstico temprano. También se realizan, en algunos casos, pruebas genéticas de una muestra sanguínea, así como pruebas de audición y acudir al oftalmólogo para detectar cualquier síntoma de cataratas u otros problemas oculares que puedan llevar a la pérdida de la vista.
TRATAMIENTO
NF1: No hay cura para la NF1 pero se pueden tratar algunos de sus efectos. Se pueden extirpar los tumores de la piel que causan dolor o desfiguración mediante cirugía, pero estes con frecuencia vuelven a crecer.NF2: Tampoco existe ninguna cura para este tipo pero se puede realizar cirugía y radioterapia para ayudar a controlar los síntomas. Las resonancias magnéticas pueden detectar pequeños tumores, lo que permite un tratamiento temprano. En algunos casos la extirpación de parte de un tumor seguida de radioterapia puede ayudar a aliviar los síntomas.
LA HISTORIA DE HUANG CHUNCAI, EL HOBRE ELEFANTE
Huang Chuncai sufre desde pequeño un neurofibroma en la
 cara, un tumor que crece poco a poco sin parar. Hasta los 31 años que tiene actualmente tuvo que vivir con un bulto en la cara que crecía y crecía hasta llegar a pesar en la actualidad 15 kgs. Desde pequeño sufrió el desprecio de sus compañeros, por lo que abandonó la escuela cuando tenía 10n años. Desde entonces vivió encerrado. Él afirma que no tuvo tan sólo un día feliz en su vida. Pero ahora su vida puede cambiar al someterse a una cirugía. Los especialistas aseguran que se trata de un caso complicado porque el tumor es enorme y hay demasiados vasos sanguíneos dentro, lo que hace que la operación sea muy difícil y si no pueden parar la sangre durante la intervención Huang morirá. Pero a pesar del riesgo Huang está decidido a sacrificarse porque asegura que nunca ha podido disfrutar de un día feliz.
cara, un tumor que crece poco a poco sin parar. Hasta los 31 años que tiene actualmente tuvo que vivir con un bulto en la cara que crecía y crecía hasta llegar a pesar en la actualidad 15 kgs. Desde pequeño sufrió el desprecio de sus compañeros, por lo que abandonó la escuela cuando tenía 10n años. Desde entonces vivió encerrado. Él afirma que no tuvo tan sólo un día feliz en su vida. Pero ahora su vida puede cambiar al someterse a una cirugía. Los especialistas aseguran que se trata de un caso complicado porque el tumor es enorme y hay demasiados vasos sanguíneos dentro, lo que hace que la operación sea muy difícil y si no pueden parar la sangre durante la intervención Huang morirá. Pero a pesar del riesgo Huang está decidido a sacrificarse porque asegura que nunca ha podido disfrutar de un día feliz. Leer artículo completo











































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}